UPGMA (unweighted pair group method with arithmetic mean)是一种相对简单的层次聚类方法。这个方法存在另一种变体 WPGMA。这个方法的创始人被认为是Sokal和Michener 。 [1]
UPGMA 演法构建出一棵有根树(树状图)表现相似矩阵或相异矩阵中的特征与结构。在算法里的每一步,距离最近的两个集群(子树)将被组合成一个更高级别的集群。任意两个集群 和
和 之间的距离,是由所有里的
之间的距离,是由所有里的 元素和所有里的
元素和所有里的 元素的距离
元素的距离 的平均值,即每个集群的元素之间的平均距离,其中
的平均值,即每个集群的元素之间的平均距离,其中  和
和 是该两个集群的基数(集合大小):
是该两个集群的基数(集合大小):

换句话说,在每一次组合成新集群的步骤中,可以由 和
和 的加权平均给出集群
的加权平均给出集群 和一个新集群
和一个新集群 之间的距离:
之间的距离:

UPGMA 算法生成的有根树状图是一个超度量树,该树需要套用等速率的假设,也就是说根到每个分支尖端的距离皆相等。当尖端是同时采样的分子数据(即DNA 、 RNA和蛋白质)时,超度量假设就等同于分子钟假设。
这个示例是基于JC69基因距离矩阵,该矩阵是根据五种细菌的5S 核糖体 RNA序列计算出来的,五种细菌如下所列[2] [3]:
枯草杆菌 Bacillus subtilis(  )
)
嗜热脂肪芽孢杆菌 <i>Bacillus stearothermophilus</i>(  )
)
魏斯氏菌 Lactobacillus viridescens(  )
)
无原枯草杆菌 Acholeplasma modicum(  )
)
藤黄微球菌 <i>Micrococcus luteus</i>(  )
)
假设有五个物件 和他们之间的相异矩阵
和他们之间的相异矩阵 :
:
|
|
a
|
b
|
c
|
d
|
e
|
| a
|
0
|
17
|
21
|
31
|
23
|
| b
|
17
|
0
|
30
|
34
|
21
|
| c
|
21
|
30
|
0
|
28
|
39
|
| d
|
31
|
34
|
28
|
0
|
43
|
| e
|
23
|
21
|
39
|
43
|
0
|
在这里, 是最小值,所以将和集群。
是最小值,所以将和集群。
令 表示现在 和 的祖先。为了让 和 与 等距,假设
表示现在 和 的祖先。为了让 和 与 等距,假设 ,这对应到了超度量的假设。在这个范例中:
,这对应到了超度量的假设。在这个范例中:
然后将更新成一个新的距离矩阵 (计算在下方),由于和的集群,该矩阵的尺寸减少了一行一列。(中粗体表示的值是由加权平均计算出的新距离)
(计算在下方),由于和的集群,该矩阵的尺寸减少了一行一列。(中粗体表示的值是由加权平均计算出的新距离)



中的斜体值不受矩阵更新影响,因为他们与第一个集群中的元素完全美有关连。
现在重复前面的三个步骤,并从新的相异矩阵开始
|
|
(a,b)
|
c
|
d
|
e
|
| (a,b)
|
0
|
25.5
|
32.5
|
22
|
| c
|
25.5
|
0
|
28
|
39
|
| d
|
32.5
|
28
|
0
|
43
|
| e
|
22
|
39
|
43
|
0
|
在这个矩阵中,  是中的最小值,所以将
是中的最小值,所以将 和元素集成新群。
和元素集成新群。
令 表示节点和的祖先。由超度量假设可以得到
表示节点和的祖先。由超度量假设可以得到 三顶点到的距离相等,即:
三顶点到的距离相等,即: ,从而可以计算出到的距离
,从而可以计算出到的距离
然后将更新成新的距离矩阵 ,数值计算如下:
,数值计算如下:


重复上述动作可以得到是
|
|
((a,b),e)
|
c
|
d
|
| ((a,b),e)
|
0
|
30
|
36
|
| c
|
30
|
0
|
28
|
| d
|
36
|
28
|
0
|
 是:
是:
|
|
((a,b),e)
|
(c,d)
|
| ((a,b),e)
|
0
|
33
|
| (c,d)
|
33
|
0
|
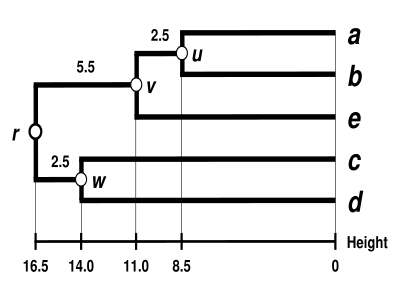
这里显示了完成的树状图。[4]它是超度量的,所有尖端( 到 ) 与 等距离 :
等距离 :

这个树状图的根是它最深的节点 。
构建 UPGMA 树的算法有 时间复杂度。使用一个堆维护两个即群之间的距离可以使时间达到
时间复杂度。使用一个堆维护两个即群之间的距离可以使时间达到 . 另外Fionn Murtagh 提出了一个
. 另外Fionn Murtagh 提出了一个 时空复杂度的算法。 [5]
时空复杂度的算法。 [5]
- ^ Sokal, Michener. A statistical method for evaluating systematic relationships. University of Kansas Science Bulletin. 1958, 38: 1409–1438.
- ^ Erdmann VA, Wolters J. Collection of published 5S, 5.8S and 4.5S ribosomal RNA sequences. Nucleic Acids Research. 1986,. 14 Suppl (Suppl): r1–59. PMC 341310
 . PMID 2422630. doi:10.1093/nar/14.suppl.r1.
. PMID 2422630. doi:10.1093/nar/14.suppl.r1.
- ^ Olsen GJ. Phylogenetic analysis using ribosomal RNA. Methods in Enzymology. 1988, 164: 793–812. PMID 3241556. doi:10.1016/s0076-6879(88)64084-5.
- ^ Swofford DL, Olsen GJ, Waddell PJ, Hillis DM. Hillis , 编. Phylogenetic inference. Sunderland, MA: Sinauer. 1996: 407–514. ISBN 9780878932825.
- ^ Murtagh F. Complexities of Hierarchic Clustering Algorithms: the state of the art. Computational Statistics Quarterly. 1984, 1: 101–113.

